Analysis Workflow in R¶
xqubit is a Python package typically used in Python environments such as Jupyter notebooks or standalone scripts. However, gene expression data are often preprocessed in R using packages such as DESeq2 or edgeR. For this reason, this tutorial demonstrates how to use xqubit from R via an interface to Python.
We present an example workflow for constructing gene co-expression networks using xqubit, based on a time-series RNA-seq dataset from Arabidopsis thaliana collected after germination, originally generated to study flower initiation [1].
The analysis consists of five main steps:
Data preparation
Quantum state encoding
Fidelity calculation
Network construction
Visualization
Data Preparation¶
xqubit requires the following inputs:
A normalized gene expression matrix
An experimental design data frame with
condition,timepoint, andreplicatecolumnsGene identifiers (optional)
Expression data should be organized as a matrix with genes as rows and samples as columns. Assuming raw count data are stored in a tab-delimited file, they can be loaded into R as follows:
x <- read.table("ath.counts.txt", header = TRUE, sep = "\t", row.names = 1)
head(x)
## T07_1 T07_2 T08_1 T08_2 T09_1 T09_2 T10_1 T10_2 T11_1 T11_2 T12_1 ...
## AT1G01010 89 183 57 123 60 75 224 105 62 86 98 ...
## AT1G01020 271 128 167 368 184 170 718 478 126 189 228 ...
## AT1G01030 34 2 20 30 11 14 17 37 1 10 30 ...
## AT1G01040 1021 645 653 1196 611 924 2474 1941 720 410 1036 ...
## AT1G01046 0 0 0 0 0 0 0 0 0 0 0 ...
## AT1G01050 1561 3334 1452 1651 1300 1224 1977 1403 2388 1864 1111 ...
As column names of the expression matrix encode experimental conditions
and replicates (condition_replicate),
we create the experimental design table by parsing the column names:
x_design <- data.frame(
sample = colnames(x),
condition = sapply(strsplit(colnames(x), "_"), `[`, 1),
replicate = sapply(strsplit(colnames(x), "_"), `[`, 2),
timepoint = as.integer(gsub("T", "", sapply(strsplit(colnames(x), "_"), `[`, 1)))
)
x_design
## sample condition replicate timepoint
## 1 T07_1 T07 1 7
## 2 T07_2 T07 2 7
## 3 T08_1 T08 1 8
## 4 T08_2 T08 2 8
## 5 T09_1 T09 1 9
## ...
Raw read counts often contain technical biases that can obscure biological signals. Normalization and gene filtering are therefore critical preprocessing steps before constructing networks.
Here we apply variance stabilizing transformation (VST), implemented in DESeq2 [2], for raw counts. One can use other normalization methods as well, such as regularized log transformation of TPM values or log transformed TMM counts.
library(DESeq2)
dds <- DESeqDataSetFromMatrix(x, x_design, design = ~ condition)
dds <- estimateSizeFactors(dds)
x_norm <- assay(vst(dds, blind = TRUE))
Since invariant or lowly expressed genes can generate spurious similarities, genes with low expression or low variability should be removed. The thresholds used below are example values and should be adjusted depending on the characteristics of the dataset.
f1 <- (rowSums(x_norm >= 5) >= (ncol(x_norm) / 3))
f2 <- (apply(x_norm, 1, mad) > 0.5)
x_norm <- x_norm[f1 & f2, ]
The resulting matrix x_norm contains normalized expression values
for genes with sufficient expression and variability,
ready for downstream analysis with xqubit.
dim(x_norm)
## [1] 1156 20
head(x_norm)
## T07_1 T07_2 T08_1 T08_2 T09_1 T09_2 T10_1 ...
## AT1G01070 10.813749 11.768753 11.404468 11.101382 11.188867 10.918834 8.869768 ...
## AT1G01110 8.806264 9.565552 8.780190 8.890654 8.613121 9.180422 9.256020 ...
## AT1G01170 10.051471 9.732266 10.286921 9.787186 10.216392 9.812231 9.358475 ...
## AT1G01190 10.285683 9.978618 10.705898 10.702856 10.268512 10.328980 8.509345 ...
## AT1G01470 10.144925 9.706032 10.369087 10.252375 10.426127 10.194991 8.499688 ...
## AT1G01580 7.961483 8.341188 7.890097 8.035285 8.010770 8.045538 7.551140 ...
Quantum State Encoding¶
After preparing the data, gene expression profiles are embedded into quantum states using xqubit. Because xqubit is implemented in Python, we access it from R via the reticulate package, which provides an interface for calling Python functions from within R.
First, load reticulate and specify the Python environment where xqubit is installed:
library(reticulate)
use_python("/bin/python", required = TRUE) # adjust path as needed
Next, import the xqubit module in R
and create a xqubit.ExpressionData object
using the normalized expression matrix
and the experimental design data frame.
xqubit <- import("xqubit")
d <- xqubit$ExpressionData(as.matrix(x_norm), x_design, rownames(x_norm))
Then, build quantum-state embeddings.
psi <- xqubit$qstate$build(d, encoding='TDP')
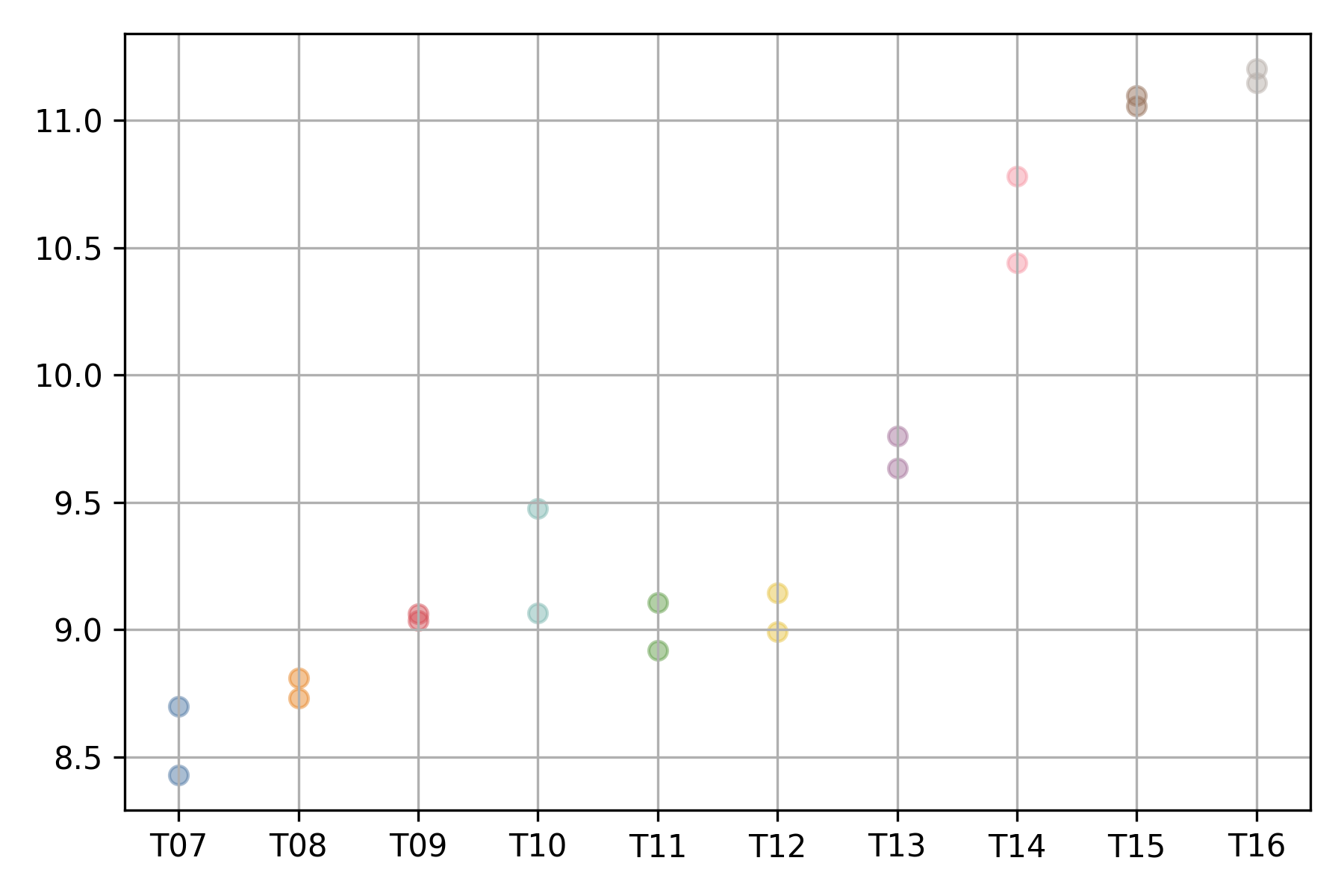
The expression profile of each gene can be visualized using
the xqubit.ExpressionData.plot method,
where the x-axis represents conditions (time points)
and the y-axis represents normalized expression values.
d$plot('AT5G61850', file_name='AT5G61850.exp.png')
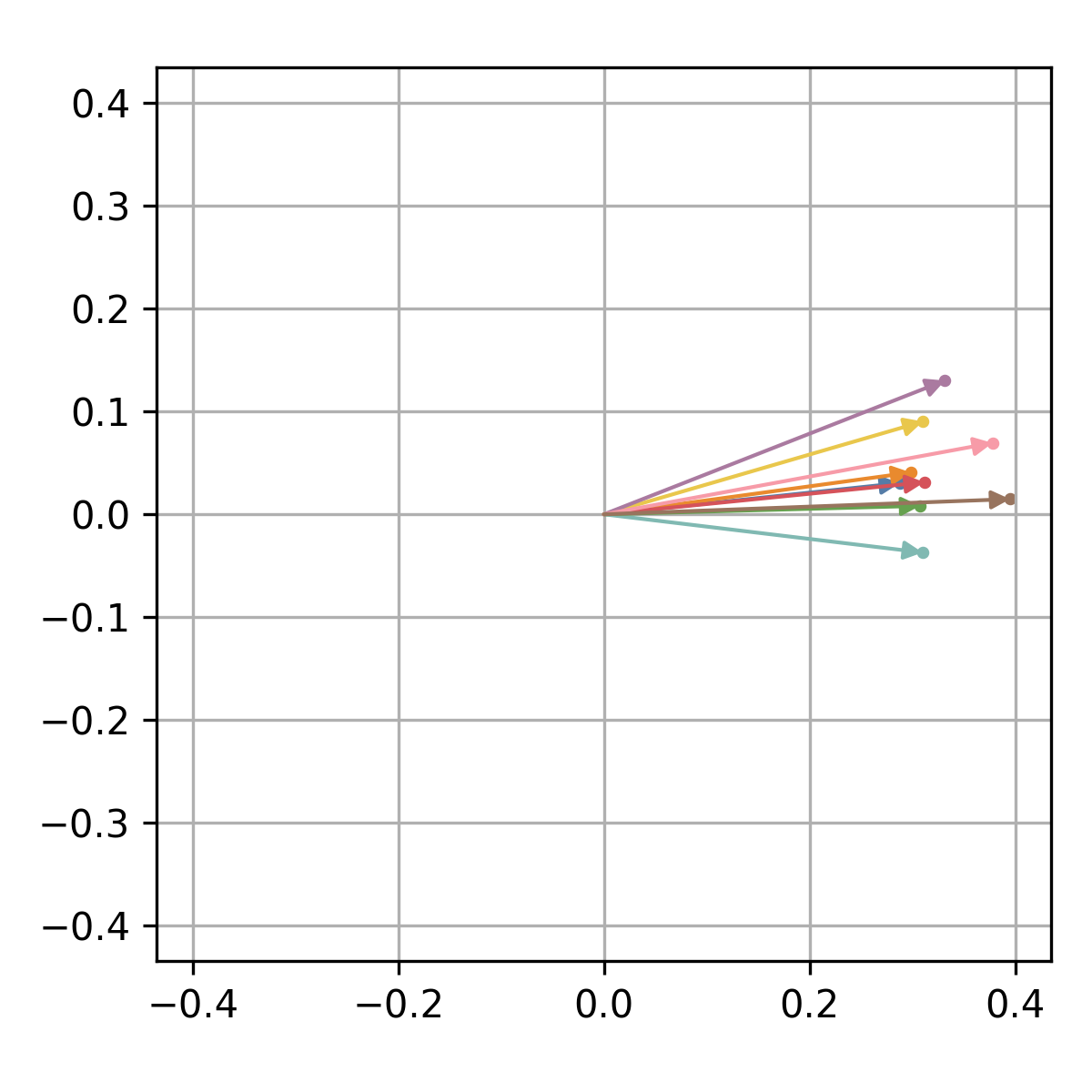
The corresponding quantum state can be visualized in the complex plane
using the xqubit.qstate.plot method.
idx <- d$genes('AT5G61850')
xqubit$qstate$plot(psi, idx, file_name='AT5G61850.qstate.png')
Fidelity Calculation¶
Fidelity between genes is calculated using the xqubit.metrics.calc_fidelity function,
which takes the psi as input and returns a fidelity matrix
where rows and columns correspond to genes and values represent pairwise similarity.
fmat <- xqubit$metrics$calc_fidelity(psi)
dim(fmat)
## [1] 1156 1156
head(fmat)
## [,1] [,2] [,3] [,4] [,5] [,6] [,7] ...
## [1,] 1.0000000 0.8027602 0.7253503 0.9887822 0.9910231 0.9532281 0.8772183 ...
## [2,] 0.8027602 1.0000000 0.5417154 0.7918268 0.7628359 0.9059334 0.9328506 ...
## [3,] 0.7253503 0.5417154 1.0000000 0.6901691 0.7442489 0.6326834 0.7284880 ...
## [4,] 0.9887822 0.7918268 0.6901691 1.0000000 0.9875654 0.9556262 0.8796378 ...
## [5,] 0.9910231 0.7628359 0.7442489 0.9875654 1.0000000 0.9366521 0.8680249 ...
## [6,] 0.9532281 0.9059334 0.6326834 0.9556262 0.9366521 1.0000000 0.9344884 ...
Network Construction¶
To construct a gene-gene network based on fidelity similarities,
we use functions from the xqubit.nx module, which implements
a flexible framework for building and analyzing networks.
There are three parameters that control network construction and community detection:
f: minimum fidelity value required for a gene-gene connectionk: number of nearest neighbors retained per generesolution: resolution parameter for Leiden community detection
To identify suitable parameter values, a grid search can be performed.
The xqubit.nx.scan_network_params method evaluates each network
based on modularity, stability, number of detected communities, and other metrics.
nx_params <- xqubit$nx$scan_network_params(
fmat,
s_cutoffs = c(0.6, 0.7, 0.8, 0.9),
k_cutoffs = c(10, 20, 30),
resolutions = c(1.0, 1.5))
The xqubit.nx.rank_network_params function ranks parameter combinations
based on predefined acceptance ranges and Pareto optimality with
respect to selected evaluation metrics.
Using the default acceptance ranges and optimization variables,
we can inspect the ranked parameter combinations as follows:
nx_params <- xqubit$nx$rank_network_params(nx_params)
nx_params <- data.frame(nx_params$to_dict(orient='list'))
best_params <- nx_params[1, ]
Note that user can customize the acceptance ranges and optimization variables
by passing the opt_vars argument to xqubit.nx.rank_network_params.
Using the selected parameters, the fidelity network is constructed and communities are detected:
nx <- xqubit$nx$build_network(fmat, s = best_params$s, k = best_params$k)
cl <- xqubit$nx$detect_communities(nx, resolution = best_params$resolution)
cl is a vector of community labels for each gene,
where labels start from 0 and genes sharing the same label belong to the same community.
Genes with no connections, as well as those in small communities that do not meet the minimum size requirement,
are assigned to community 0.
Thus, community 0 is typically excluded from downstream analyses.
table(cl)
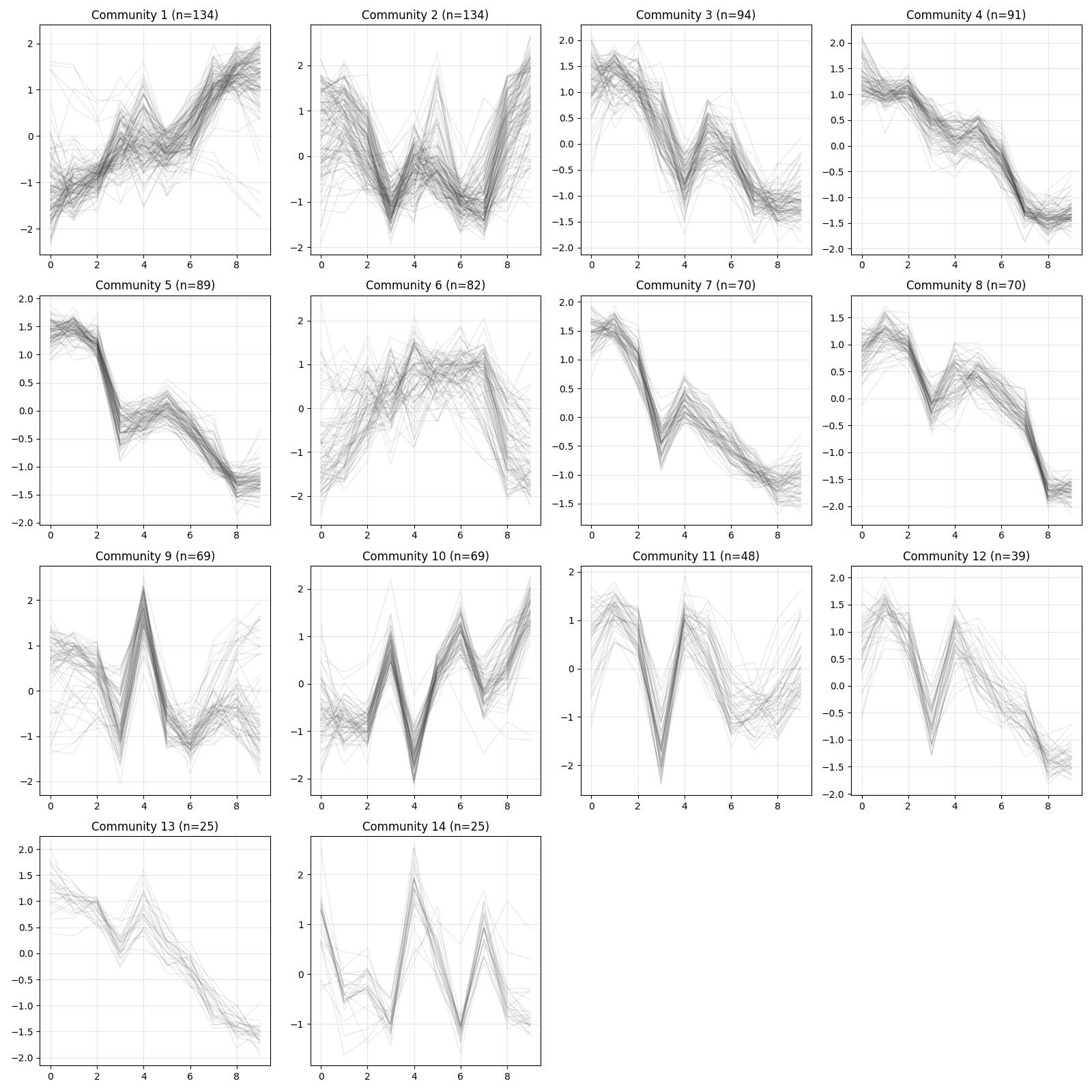
## 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
## 117 134 134 94 91 89 82 70 70 69 69 48 39 25 25
Communities can also be saved to a file:
comms_df <- data.frame(gene = rownames(x_norm), community = cl)
write.table(comms_df, "communities.txt",
sep = "\t", row.names = FALSE, col.names = TRUE, quote = FALSE)
Visualization¶
Visualization utilities are available for inspecting expression patterns within communities.
z <- d$exp(avg_replicates = TRUE, zscore = TRUE)
xqubit$nx$plot_communities(cl, z, file_name = "comm_expression.png")