Analysis Workflow in Python¶
xqubit is a Python package designed for use in Python environments such as Jupyter notebooks or standalone scripts. To use xqubit, users should prepare gene expression data (i.e., normalized expression values) in a format suitable for analysis in Python.
This tutorial presents a standard workflow for constructing gene co-expression networks using xqubit, based on a time-series RNA-seq dataset from Arabidopsis thaliana collected after germination, originally generated to study flower initiation [1].
The analysis consists of five main steps:
Data preparation
Quantum state encoding
Fidelity calculation
Network construction
Visualization
Data Preparation¶
xqubit requires the following inputs:
A normalized gene expression matrix
An experimental design data frame with
condition,timepoint, andreplicatecolumnsGene identifiers (optional)
Expression data should be organized as a matrix with genes as rows and samples as columns. Assuming normalized expression data are stored in a tab-delimited file, they can be loaded into Python as follows:
Note
Raw read counts often contain technical biases that can obscure biological signals. Normalization and gene filtering are therefore critical preprocessing steps before constructing networks.
Since invariant or lowly expressed genes can generate spurious similarities, such genes should be removed. In practice, preprocessing steps such as normalization and gene filtering are typically performed in R using packages such as DESeq2 or edgeR, which provide well-established analysis frameworks.
For this reason, we recommend preparing the data in R and then importing the processed data into Python for downstream analysis with xqubit.
import pandas as pd
x = pd.read_csv("ath.vst.txt", sep="\t", index_col=0)
x.head()
## T07_1 T07_2 T08_1 T08_2 T09_1 T09_2 T10_1 T10_2 T11_1 T11_2 T12_1 T12_2 T13_1 T13_2 T14_1 T14_2 T15_1 T15_2 T16_1 T16_2
## gene
## AT1G01070 10.68 11.70 11.32 10.99 11.09 10.79 8.35 8.69 9.20 8.28 9.23 8.72 8.28 9.05 7.92 7.24 5.20 5.13 5.12 4.72
## AT1G01110 8.26 9.24 8.22 8.37 7.99 8.76 8.86 8.97 8.33 8.59 8.70 8.80 9.16 9.06 9.32 9.61 9.48 9.49 9.45 9.54
## AT1G01170 9.82 9.45 10.09 9.51 10.01 9.54 8.99 8.98 10.54 10.76 9.81 9.50 9.35 9.23 9.88 9.68 10.32 10.38 10.64 10.81
## AT1G01190 10.09 9.74 10.56 10.56 10.07 10.14 7.83 8.19 7.10 8.03 8.40 8.09 7.75 8.38 6.97 6.37 5.20 5.01 5.00 5.35
## AT1G01470 9.93 9.42 10.19 10.05 10.25 9.99 7.82 8.00 8.70 7.98 8.99 8.27 7.62 8.40 7.33 6.90 5.74 5.74 5.75 5.60
As column names of the expression matrix encode experimental conditions
and replicates (condition_replicate),
we create the experimental design table by parsing the column names:
x_design = pd.DataFrame({
"sample": x.columns,
"condition": [col.split("_")[0] for col in x.columns],
"replicate": [col.split("_")[1] for col in x.columns],
"timepoint": [int(col.split("_")[0][1:]) for col in x.columns]
})
x_design
## sample condition replicate timepoint
## 0 T07_1 T07 1 7
## 1 T07_2 T07 2 7
## 2 T08_1 T08 1 8
## 3 T08_2 T08 2 8
## 4 T09_1 T09 1 9
## ...
gene = x.index.tolist()
## ['AT1G01070', 'AT1G01110', 'AT1G01170', 'AT1G01190', 'AT1G01470', ...]
Quantum State Encoding¶
After preparing the data, gene expression profiles are encoded into quantum states using xqubit.
First, import the xqubit module and create a xqubit.ExpressionData object using the expression matrix and the experimental design data.
import xqubit
d = xqubit.ExpressionData(x.values, x_design, gene)
Next, construct quantum-state representations of gene expression profiles.
psi = xqubit.qstate.build(d, encoding='TDP')
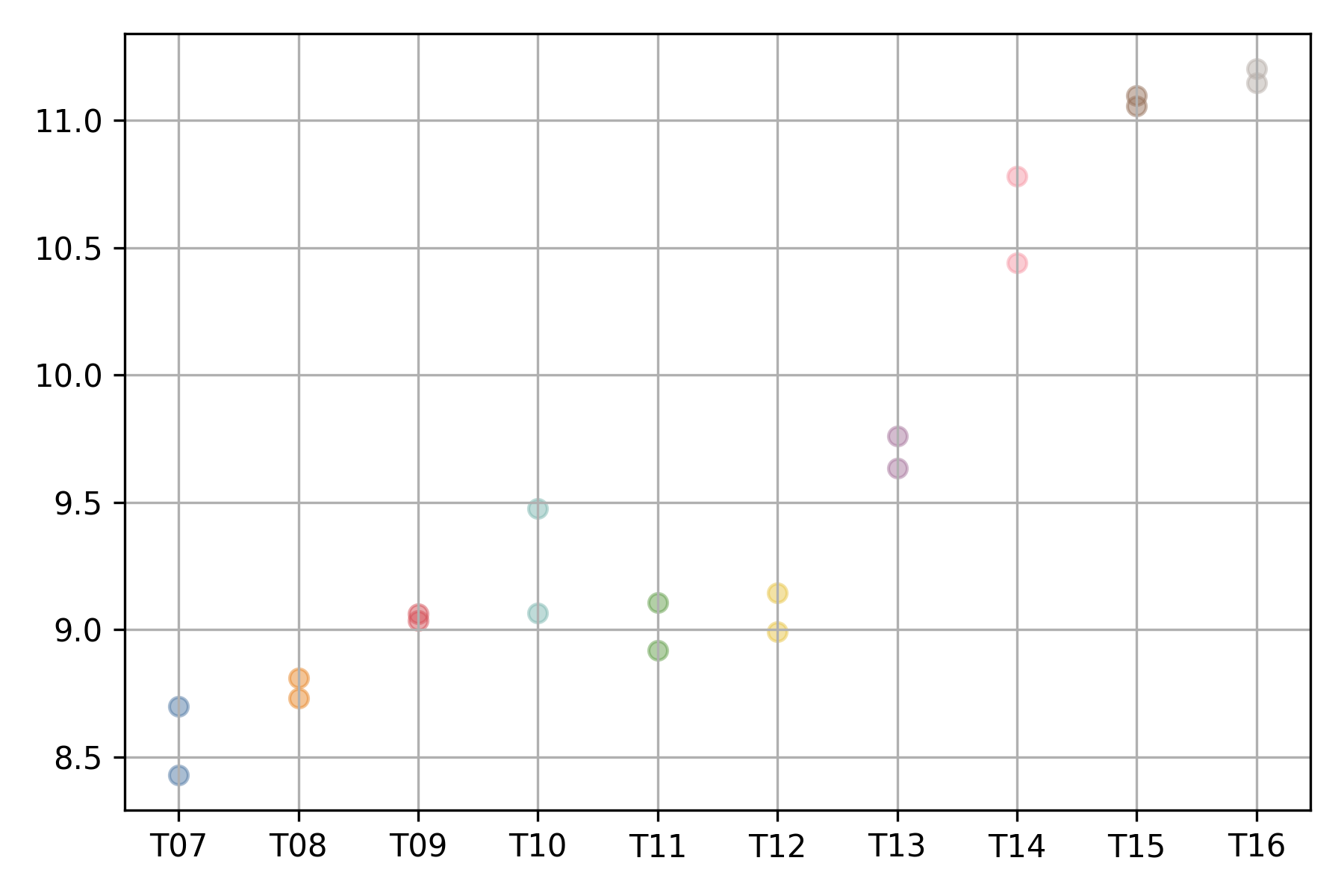
The expression profile of each gene can be visualized using
the xqubit.ExpressionData.plot_exp method,
where the x-axis represents conditions (time points)
and the y-axis represents normalized expression values.
d.plot('AT5G61850', file_name='AT5G61850.exp.png')
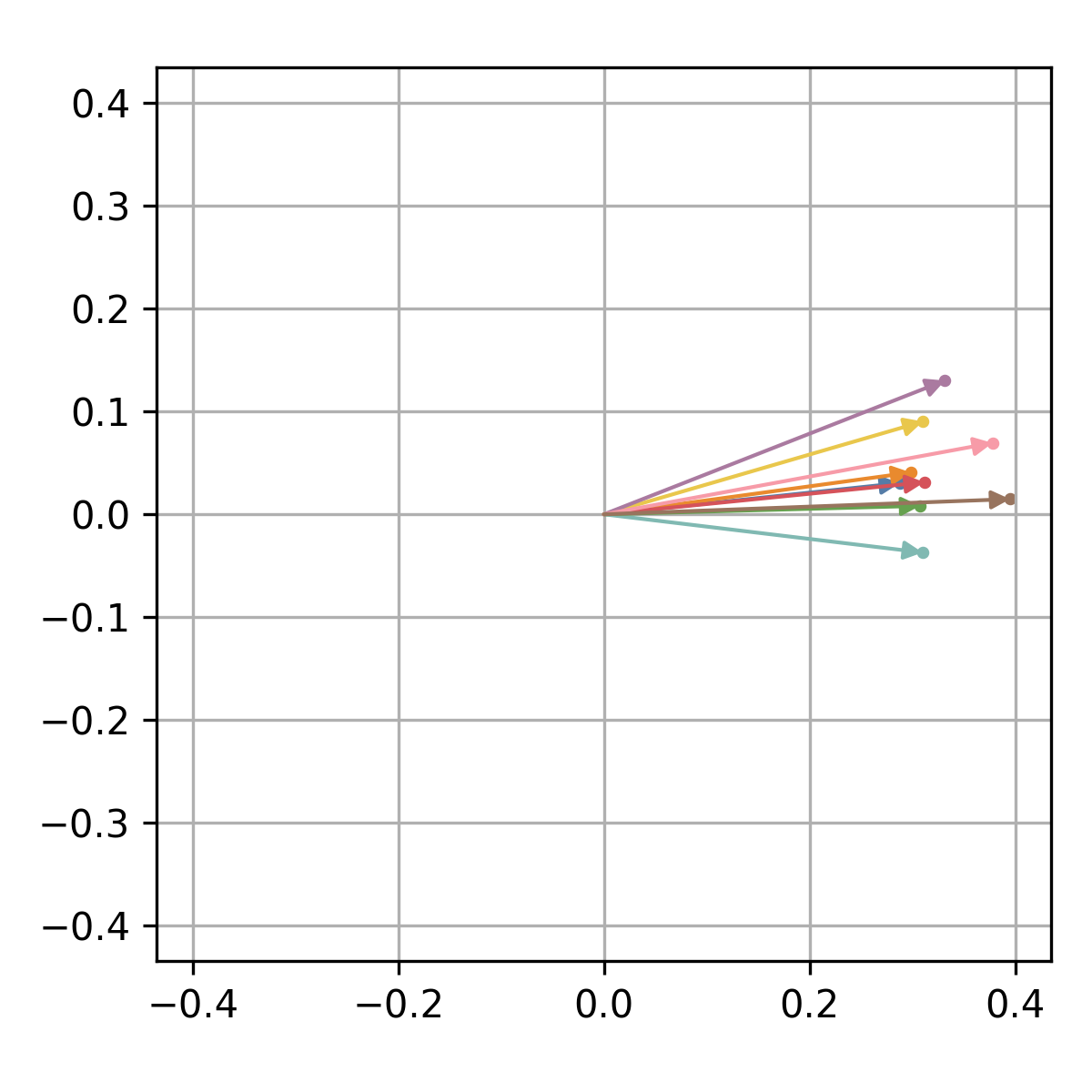
The corresponding quantum state can be visualized in the complex plane
using the xqubit.qstate.plot method.
idx = d.genes('AT5G61850')
xqubit.qstate.plot(psi, idx, file_name='AT5G61850.qstate.png')
Fidelity Calculation¶
Fidelity between genes is computed using the xqubit.metrics.calc_fidelity function.
This function takes the psi as input and returns a matrix in which rows and columns correspond to genes, and each entry represents the pairwise similarity between gene-specific quantum states.
fmat = xqubit.metrics.calc_fidelity(psi)
fmat.shape
## (1156, 1156)
fmat[:5, :5]
## array([[1. , 0.77728261, 0.68946785, 0.99036295, 0.99065266],
## [0.77728261, 1. , 0.53816368, 0.7698045 , 0.75199344],
## [0.68946785, 0.53816368, 1. , 0.66055549, 0.72229802],
## [0.99036295, 0.7698045 , 0.66055549, 1. , 0.98765317],
## [0.99065266, 0.75199344, 0.72229802, 0.98765317, 1. ]])
Network Construction¶
To construct a gene–gene network based on fidelity similarities, we use functions from the xqubit.nx module, which provides utilities for network construction and analysis.
Three parameters control network construction and community detection:
f: minimum fidelity threshold for defining an edge between genesk: number of nearest neighbors retained per generesolution: resolution parameter for Leiden community detection
To identify suitable parameter values, a grid search can be performed.
The xqubit.nx.scan_network_params method evaluates each parameter
combination using metrics such as modularity, stability, and the number of detected communities.
nx_params = xqubit.nx.scan_network_params(
fmat,
s_cutoffs = [0.6, 0.7, 0.8, 0.9],
k_cutoffs = [10, 20, 30],
resolutions = [1.0, 1.5])
The xqubit.nx.rank_network_params function ranks parameter combinations
based on predefined acceptance ranges and Pareto optimality with
respect to selected evaluation metrics.
- Using the default settings,
the top-ranked parameter combination can be retrieved as follows:
nx_params = xqubit.nx.rank_network_params(nx_params)
best_params = nx_params.iloc[0]
Note that user can customize the acceptance ranges and optimization variables
by passing the opt_vars argument to xqubit.nx.rank_network_params.
Using the selected parameters, the network is constructed and communities are detected:
nx = xqubit.nx.build_network(fmat, s = best_params.s, k = best_params.k)
cl = xqubit.nx.detect_communities(nx, resolution = best_params.resolution)
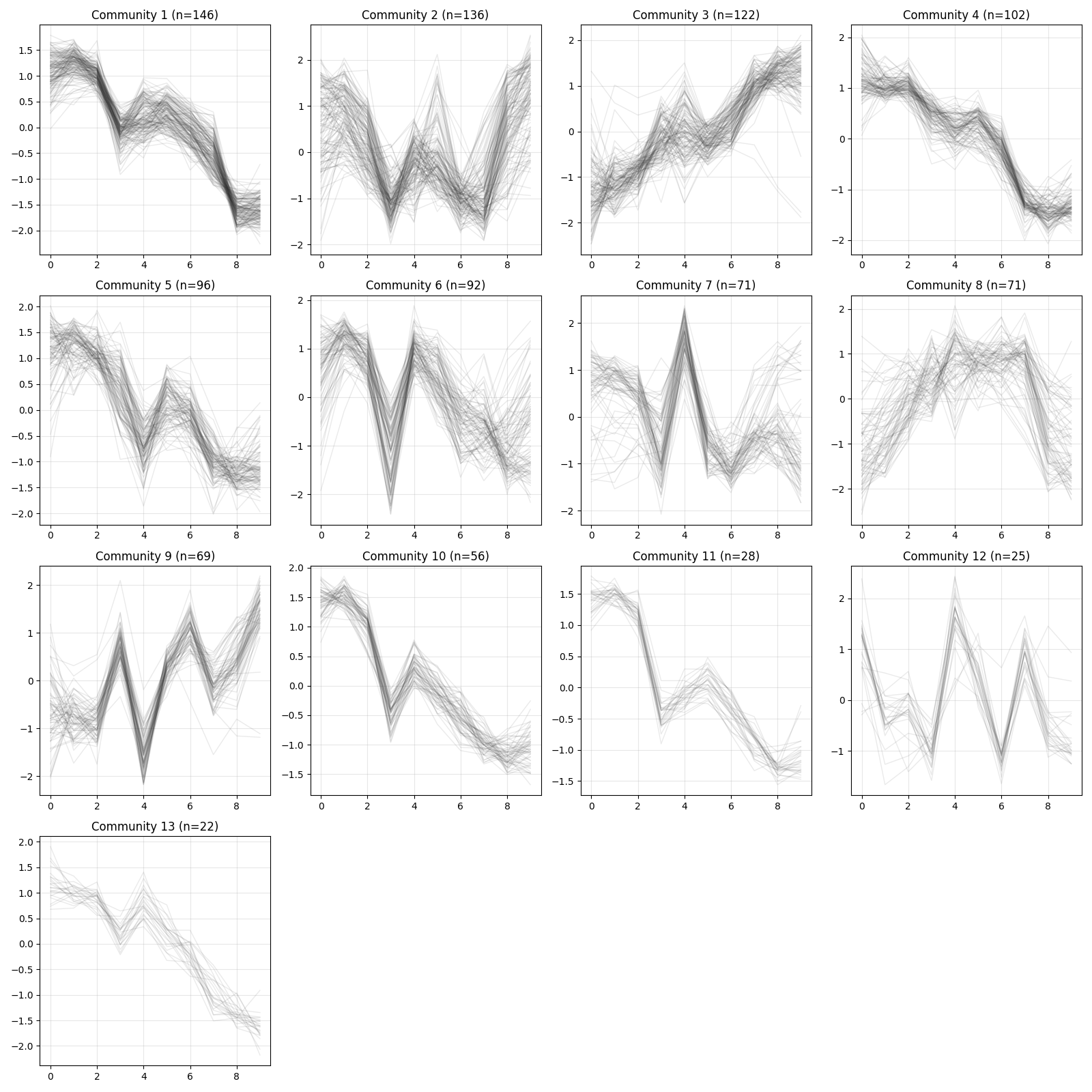
cl is a list of community labels for each gene, where labels start from 0 and genes sharing the same label belong to the same community.
Genes with no connections, as well as those in small communities that do not meet the minimum size requirement, are assigned to community 0.
Community 0 is typically excluded from downstream analyses.
pd.Series(cl).value_counts()
## 1 146
## 2 136
## 3 122
## 0 120
## 4 102
## 5 96
## 6 92
## 7 71
## 8 71
## 9 69
## 10 56
## 11 28
## 12 25
## 13 22
## Name: count, dtype: int64
The detected communities can also be saved to a file:
comms_df = pd.DataFrame({
"gene": d.genes(),
"community": cl
})
comms_df.to_csv("communities.txt", sep="\t", index=False)
Visualization¶
Visualization utilities are available for inspecting expression patterns within detected communities.
z = d.exp(avg_replicates=True, zscore=True)
xqubit.nx.plot_communities(cl, z, file_name = "comm_expression.png")